Balding, Petrovski & Silk - Genome Research
Predicting disease-potential: epilepsy genes
Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation
J Traynelis, M Silk, Q Wang, S Berkovic, L Liu, D Ascher, D Balding and S Petrovski, Genome Research (2017) doi:10.1101/gr.226589.117
Large samples of human standing variation data highlight regional variation in the tolerance to missense variation within the protein-coding sequence of genes. This information is not well captured by existing bioinformatic tools, but is effective in improving variant interpretation.
David Balding, Associate Member Slavé Petrovski and their co-supervised PhD student, Michael Silk address this limitation in exisiting tools in their recently published Genome Research paper, where they introduce the missense tolerance ratio (MTR), which summarizes available human standing variation data within genes to encapsulate population level genetic variation.
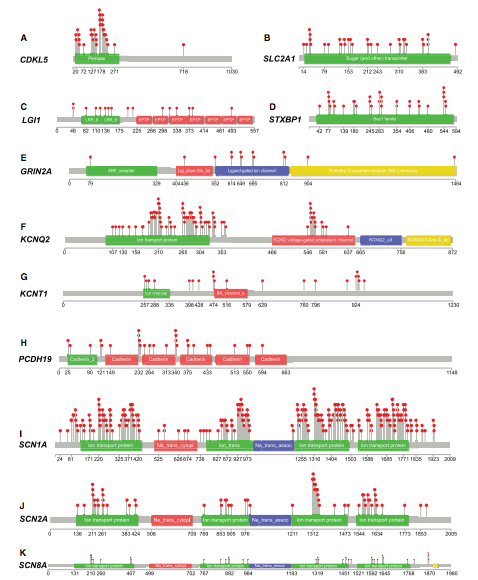
They found that patient-ascertained pathogenic variants preferentially cluster in low MTR regions (P < 0.005) of well-informed genes. By evaluating 20 publicly available predictive tools across genes linked to epilepsy, they also highlighted the importance of understanding the empirical null distribution of existing prediction tools, as these vary across genes. Subsequently integrating the MTR with the empirically selected bioinformatic tools in a gene-specific approach demonstrates a clear improvement in the ability to predict pathogenic missense variants from background missense variation in disease genes.
Whilst the focus on this study was on the application to epilepsy genes; the framework is applicable to disease genes beyond epilepsy.
Figure 2 (taken from paper): The distribution of the 606 qualified pathogenic variants (red circles) among the 11 epilepsy genes, The distribution of the 606 qualified pathogenic variants across genes.